写在前面的话
本系列将基于张泽民团队发表的TNBC免疫图谱进行挖掘(Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer: Cancer Cell00499-2)),旨在探索原发灶和转移灶免疫微环境的差异及其原因。
准确严谨的采集和清洗数据是单细胞分析的开始,数据本身的质量和采集过程的规范程度决定了研究结果的可靠性。
样本信息
患者来源
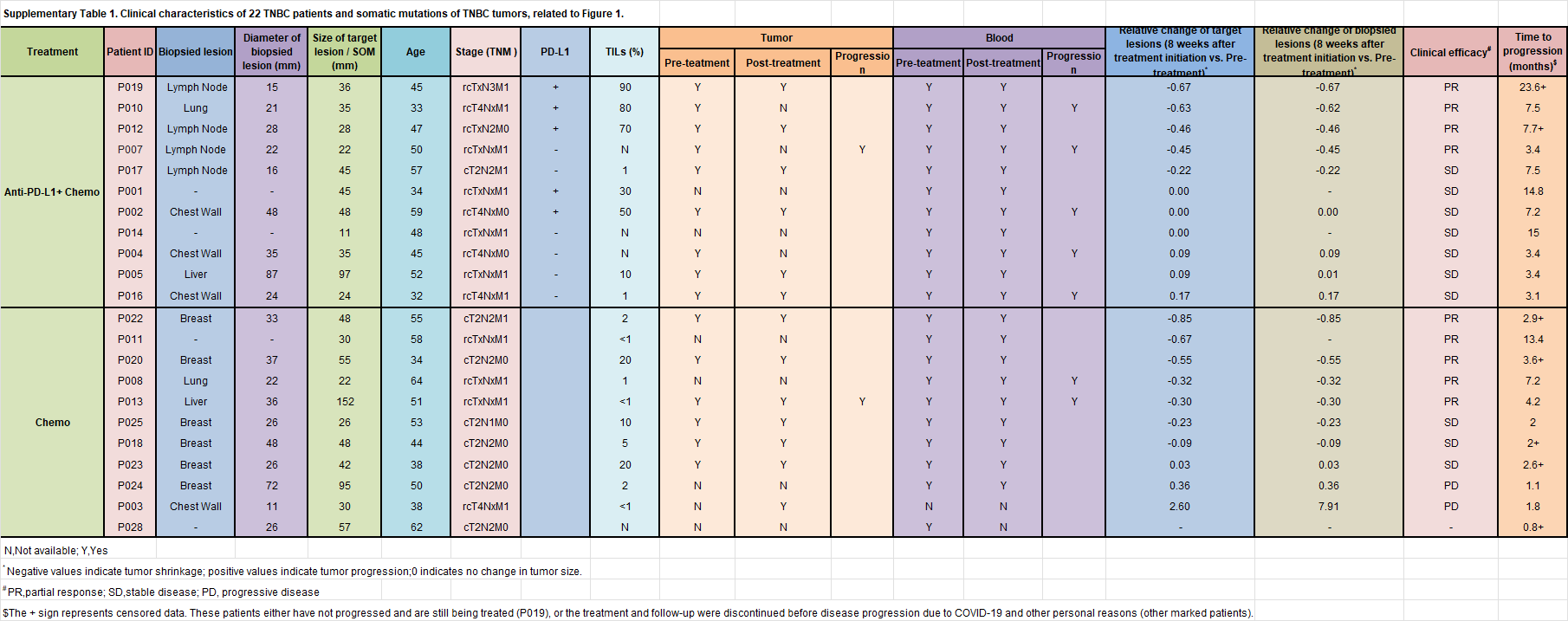
国家癌症中心开发的DTHealth TrakCare系统中记录了患者信息,纳入本研究的22名患者满足:(1)组织学确诊晚期TNBC;(2)既往没有化疗、靶向及免疫等全身治疗;(3)研究中接受紫杉醇单药治疗或紫杉醇加阿替利珠单抗治疗。
患者信息
- 本研究所有患者均被诊断为晚期TNBC,大多数患者患有转移性疾病;
- 事实上,一些患者是接受新辅助治疗的局部晚期患者;
- 患者为女性,年龄在32-64岁之间,中位值为49岁;
样本收集、文库制备和测序
研究使用16针吸穿刺取乳腺、胸壁肿瘤样本;18号针取淋巴结、肝转移灶样本。测序仪器是10x Genomics公司的HiSeq X Ten,平台为GPL20795。DNA片段被打碎为150bp,进行双端测序。
单细胞转录组数据处理
10x Genomics公司提供的Cell Ranger(V 3.0.1)用于汇总原始数据,过滤低质量读段,将reads与人类参考基因组(GRCh38)比对,分配细胞Barcode,并生成UMI矩阵。值得注意的是,对于10x Genomics公司的测序,由于UMI的存在,使用Raw Count即可反映真实的基因表达,不需要使用标准化方法进行基因定量(生物信息学笔记(三):基因定量)。但注意,这里所谓“真实的基因表达”反映的是测序过程中的真实值,即不必考虑测序深度和转录本长度的值;而单细胞测序中,由于转录本的低捕获率,我们假设合格细胞基因表达总量应该类似,所以还要进行标准化排除不同细胞测序质量的差异。
原文使用Scanpy进行下游分析,质控步骤包括:
- 过滤在少于10个细胞中表达的基因;
- 过滤少于200个基因的细胞中检测到的基因;
- 保留了600-120,000个UMIs、400-8,000个基因和小于10%线粒体基因计数的高质量细胞;
- 将Scrublet应用于每个测序文库,以去除预期的双峰率为6%的潜在双峰,并滤除doubletScore大于90%分位数的细胞;
接着,我们进行标准化操作:
- 标准化每个细胞的总计数(库大小):这一步是为了消除不同细胞间样本量的差异。库大小通常是指一个细胞中检测到的总UMI数。通过将每个细胞的UMI计数除以该细胞的总UMI计数,可以确保数据在不同细胞间是可比的。
- 乘以1e6:这一步是为了放大数据,使其更易于处理。通常在生物信息学中,进行这样的转换是为了避免处理极小的数字。
- 对数据进行对数变换:最后,对标准化后的数据进行对数变换是为了减少数据中极端值的影响,使数据的分布更接近正态分布。这通常有助于后续的统计分析和可视化。
前面的步骤完成了单细胞转录组数据的质控和标准化,下面进行矩阵降维,进一步排除无关变量的影响,同时压缩分析规模:
- 选择前4000个可变基因,排除大部分背景基因的影响;
- 从标准化表达矩阵中去除总计数、线粒体基因计数和热休克蛋白(HSP)相关基因计数的变异,以避免偏差;
- 主成分分析,把4000个可变基因降为50个主成分;
- 由此,我们得到了50×细胞数的矩阵,即对于每个细胞有50个变量,使用前2个或者3个主成分进行可视化,通过标注不同样本,发现潜在的批次效应,并通过相应的算法(在R中可以通过Harmony)去除;
- 由此,我们尽可能避免了因非研究变量因素导致的系统误差,得到新的标准化矩阵。
最后,Leiden算法寻找细胞簇和分群,UMAP和TSNE进行作图,推荐使用COSG确定Marker。